- HÉMOSTASE ET HÉMORRAGIES
- HÉMOSTASE ET HÉMORRAGIESLe sang est composé d’un liquide, le plasma, et d’éléments figurés, les globules rouges, les globules blancs, les plaquettes sanguines. C’est au niveau du plasma et des plaquettes que se trouvent les mécanismes capables de transformer le sang liquide en une masse solide. Ce phénomène, nommé coagulation, s’associe à des modifications vasculaires, et l’ensemble est désigné sous le nom d’hémostase (arrêt de l’hémorragie).Le caillot sanguin résulte de la transformation du fibrinogène, substance protéique normalement en solution dans le plasma, en une forme insoluble: la fibrine. Cette réaction est provoquée par la thrombine, enzyme dont l’activité est sous la dépendance de nombreux facteurs, dont la nomenclature et la nature biochimique ont longtemps compliqué l’étude de la coagulation.Les hémorragies (effraction du sang hors des vaisseaux) s’observent dans deux circonstances bien différentes: ou bien les mécanismes de l’hémostase sont normaux, mais débordés par une atteinte locale des vaisseaux (c’est le cas des plaies artérielles), ou bien ils sont en défaut par suite d’atteintes congénitales ou acquises portant soit sur les petits vaisseaux, soit sur les plaquettes, soit sur les facteurs de coagulation. On examinera successivement les mécanismes de l’hémostase normale et les diverses affections où ils sont perturbés, entraînant un syndrome hémorragique.1. Mécanisme de l’hémostaseOn désigne sous le nom d’hémostase l’ensemble des phénomènes grâce auxquels l’organisme obtient l’arrêt d’une hémorragie. La formation d’un caillot au niveau d’une brèche vasculaire représente un mécanisme de défense indispensable à la vie; elle se distingue de la thrombose (formation de caillot intravasculaire), qui, par suite d’embolie, met la vie en danger.Pour que l’hémostase ne dépasse pas son but et n’aboutisse pas à une thrombose extensive, intervient l’équilibre très complexe entre les facteurs coagulants d’une part, les inhibiteurs de la coagulation et les facteurs de lyse (c’est-à-dire de dissolution du caillot) d’autre part.L’hémostase se déroule en plusieurs temps, qui d’ailleurs s’imbriquent: temps vasculaire et temps plaquettaire constituant l’hémostase primaire, temps plasmatique, constituant l’hémostase secondaire.Temps vasculaireLa lésion d’un vaisseau provoque une rétraction (capillaires, veinules) ou une vasoconstriction (artères), ce qui aboutit à une réduction du calibre vasculaire. Cela a pour effet de ralentir le débit sanguin et de favoriser la formation du «thrombus blanc».Temps plaquettaireLes plaquettes sanguines (ou thrombocytes) sont des corpuscules de deux à quatre microns dépourvus de noyau, mais contenant des granulations azurophiles [cf. SANG]. Elles adhèrent en quelques secondes au collagène mis à nu par la rupture de l’endothélium (couche monocellulaire non «mouillable» qui tapisse l’intérieur des vaisseaux), et s’agrègent entre elles pour former un amas qui obstrue la brèche vasculaire: c’est le «clou plaquettaire» de G. Hayem ou «thrombus blanc». Cette agrégation est due essentiellement à la libération d’ADP (adénosine diphosphate), qui provient des cellules vasculaires lésées et des plaquettes elles-mêmes. Le bouchon formé est d’abord, ainsi qu’une éponge, perméable au sang. Sa contraction, liée à l’action d’une substance contractile (appelée thrombosthénine), contenue dans les plaquettes, assure son imperméabilisation et l’arrêt momentané de la circulation.Au sein de cet amas, les plaquettes perdent leur contour: c’est la «métamorphose visqueuse»; elles libèrent alors des produits actifs tels que la sérotonine (5-hydroxytryptamine) vasoconstrictrice, et le facteur no 3 intervenant dans la coagulation.À ces notions classiques, ajoutons les acquisitions nombreuses obtenues au cours de cette dernière décennie sur l’hémostase primaire et sur les interactions plaquettes-paroi vasculaire:L’adhésion des plaquettes au collagène et du sous-endothélium nécessite la présence d’un facteur absent dans la maladie dite de Willebrand, ainsi qu’une structure normale des glycoprotéines de la membrane plaquettaire, maintenant bien définies.L’agrégation plaquettaire fait intervenir, outre l’ADP déjà mentionné, la thrombine et le système des prostaglandines .Les prostaglandines sont des acides gras insaturés donnant rapidement naissance à des dérivés de l’acide arachidonique. Deux d’entre eux jouent un rôle important dans l’hémostase primaire: le thromboxane A2, agrégant plaquettaire, qui provient de la membrane plaquettaire, la prostacycline (ou PGI2) synthétisée par l’endothélium vasculaire, qui s’oppose à l’adhésion et à l’agrégation des plaquettes à la paroi vasculaire. Ces deux substances antagonistes jouent un rôle important. C’est ainsi que les médicaments à visée antithrombotique, telle l’aspirine, inhibent la formation du thromboxane A2, tandis que la prostacycline injectée par voie intraveineuse est susceptible de s’opposer à la coagulation intravasculaire.Temps plasmatiqueLe temps plasmatique constitue la coagulation proprement dite; elle aboutit à la formation d’un «thrombus rouge» ou caillot de fibrine enserrant dans ses mailles les globules rouges. Elle fait intervenir de très nombreux facteurs numérotés de I à XIII par le Comité international de nomenclature (cf. tableau). Tous ces facteurs sont des protéines.Selon le schéma proposé par Morawitz en 1905, la fibrine se forme à partir d’un précurseur soluble, le fibrinogène, grâce à l’action de la thrombine; celle-ci dérive d’un autre précurseur, la prothrombine:
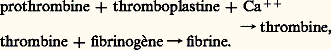 Dans ce qui suit, on étudiera brièvement les différents stades de la séquence aboutissant à la fibrine.ThromboplastinoformationLa transformation de la prothrombine en thrombine fait intervenir un ensemble très complexe d’activateurs (primitivement désignés sous le nom générique de thromboplastines). Elle résulte de la convergence de deux systèmes de thromboplastines différents, mais tous deux nécessaires. L’un fait intervenir des facteurs d’origine tissulaire: c’est la thromboplastinoformation extrinsèque, puisqu’elle met en jeu des éléments qui n’appartiennent pas au sang. L’autre se forme à partir d’éléments contenus dans le sang lui-même; c’est la thromboplastinoformation intrinsèque.La thromboplastinoformation intrinsèque est déclenchée par toute attrition vasculaire ou par le contact du sang avec une surface étrangère, tel le verre. Elle fait intervenir un «facteur contact» (XII), point de départ d’une véritable cascade de réactions enzymatiques dans lesquelles un facteur, une fois activé, active à son tour celui qui lui succède dans la chaîne enzymatique (fig. 1). On dit qu’un facteur s’est activé quand il acquiert une activité enzymatique spécifique. L’activation peut être de nature physique, telle celle du facteur Hageman par contact avec les surfaces «mouillables» (verre, silice...) ou de nature chimique, comme pour tous les autres facteurs de coagulation. En conclusion, à la suite de cette réaction en chaîne, le facteur X est activé.L’activation du facteur X a lieu simultanément dans l’organisme par thromboplastinoformation extrinsèque , mécanisme plus rapide que celui qui vient d’être énuméré. Celle-ci fait intervenir des sucs tissulaires à la place du facteur VIII activé. En d’autres termes, des extraits de tissus (en particulier de poumon, de cerveau, de placenta) accélèrent considérablement la coagulation. Ils sont libérés au niveau des blessures par suite de l’attrition tissulaire. Pour être actifs, ces sucs tissulaires doivent réagir avec un facteur normal du plasma, le facteur VII ou proconvertine (fig. 1). Le facteur X activé va s’associer à un autre facteur plasmatique, le facteur V (ou proaccélérine), qui se trouve activé par les premières traces de thrombine formée.ThrombinoformationLes facteurs X et V activés forment, en présence d’ions calcium et de micelles de lipides (phospholipides), un complexe doué d’une activité enzymatique, la prothrombinase, qui transforme la prothrombine en thrombine par scission protéolytique (fig. 1): c’est la thrombinoformation.La staphylocoagulase, provenant de certaines souches de staphylocoque doré, est également capable de se combiner directement avec la prothrombine humaine pour démasquer son site actif et coaguler le fibrinogène du sang.FibrinoformationLa thrombine une fois formée transforme le fibrinogène (facteur I) soluble en un réticulum de fibrine insoluble (caillot proprement dit). Les molécules de fibrinogène, qui sont très allongées, sont attaquées près de leur extrémité. Elles se trouvent ainsi amputées d’une charge négative (acides aminés diacides). Elles cessent de se repousser par des forces électrostatiques et se polymérisent, c’est-à-dire s’associent pour former un réseau solide.Le facteur XIII ou FSF, facteur de stabilisation de la fibrine, intervient pour consolider ce réseau.Inhibiteurs physiologiques de la coagulationComment expliquer que le sang coagule si rapidement dès qu’il est sorti des vaisseaux, alors que les coagulations intravasculaires spontanées sont rares? L’existence d’inhibiteurs de la coagulation est une des réponses à ce problème.Les facteurs de coagulation sont stables dans le plasma et se renouvellent avec une rapidité qui est propre à chaque facteur. Leur synthèse s’effectue au niveau du foie: elle est continue et équilibre la destruction physiologique de ces protéines très spécialisées. Lorsqu’on bloque la synthèse, la demi-vie (temps de destruction de la moitié de la quantité initialement en circulation) s’échelonne entre quatre heures, pour le facteur VII, et trois à quatre jours, pour le fibrinogène et la prothrombine.Dans les conditions physiologiques, une fois activés, les facteurs de coagulation sont très rapidement détruits au niveau du foie ou dans la circulation par des inhibiteurs, dont le principal est l’antithrombine III , capable de détruire progressivement de fortes quantités de thrombine non utilisées pour la coagulation du fibrinogène. L’héparine forme avec l’AT III un complexe qui accélère considérablement l’action anticoagulante.Fibrinolyse du caillotLa coagulation une fois terminée, le caillot se rétracte sous l’influence des plaquettes enserrées dans le réseau de fibrine. Ultérieurement, il va s’organiser; le réseau de fibrine prépare la cicatrisation en servant de matrice pour le développement de fibroblastes (tissu conjonctif).Un caillot non organisé est susceptible de se lyser c’est-à-dire de se dissoudre, à la suite de l’activation d’une enzyme fibrinolytique, appelée plasmine , qui dérive d’un précurseur inerte nommé plasminogène. Cette activation résulte de l’action d’activateurs tissulaires ou vasculaires, ou de l’urokinase, substance trouvée dans les urines normales (fig. 2).Certains streptocoques produisent un activateur du plasminogène nommé streptokinase (ou streptolysine).La plasmine est capable de scinder la fibrine en plusieurs fragments, qui agissent comme des inhibiteurs compétitifs de la transformation du fibrinogène en fibrine ou de la polymérisation de la fibrine. Il existe dans le plasma un inhibiteur de la plasmine à action progressive, l’antiplasmine, ainsi que l’alpha-2-macroglobuline qui a une action beaucoup plus rapide.2. HémorragiesLes hémorragies peuvent être de cause locale ou générale.Hémorragies de cause localeDans les hémorragies de cause locale, le mécanisme de l’hémostase n’est pas en cause; l’ensemble des défenses de l’organisme intervient en vue de remédier à la perte sanguine et de déterminer son arrêt. Une plaie artérielle avec issue en saccades de sang rouge vif impose souvent une intervention d’urgence: pose d’un garrot, compression de l’artère en amont, suture ou ligature chirurgicale, faute de quoi un état d’anémie aiguë se constitue très rapidement, caractérisant le choc hémorragique, avec pâleur, perte de conscience, pouls petit et rapide, effondrement de la tension artérielle.De telles hémorragies se voient non seulement dans les sections accidentelles d’artères, au cours de traumatismes de la route ou du travail, mais également dans certaines affections obstétricales (grossesse extra-utérine) où une artère peut être érodée. C’est aussi le cas des grandes hémoptysies (crachements de sang faisant irruption dans les voies aériennes), et des grandes hématémèses (vomissements de sang), qui compliquent certains ulcères d’estomac creusant en profondeur, ou certaines cirrhoses avec hypertension portale (rupture de varices œsophagiennes nécessitant la pose d’un ballon œsophagogastrique gonflable).Dans tous ces cas d’hémorragies dramatiques de cause locale, deux gestes s’imposent: l’hémostase locale (souvent chirurgicale), et la transfusion massive de sang.Hémorragies de cause généralePrincipales localisationsLes hémorragies cutanées , pétéchies (petites hémorragies cutanées de la taille de confettis, qui surviennent spontanément sous forme de «purpura»), et ecchymoses (classiques «bleus») se rencontrent aussi bien dans les purpuras d’origine vasculaire ou plaquettaire, que dans les atteintes de la coagulation proprement dite.Les hémorragies muqueuses comprennent les épistaxis (saignement de nez), les gingivorragies (saignement des gencives), les hémorragies après extractions dentaires (l’extraction dentaire révèle des tendances hémorragiques demeurées souvent latentes; en effet, il y a véritable arrachement d’un paquet vasculonerveux sans hémostase locale).Les hématuries , ou hémorragies urinaires, se rencontrent aussi bien dans les atteintes rénales que dans les syndromes hémorragiques des purpuras ou des troubles de la coagulation (hémophilie, surdosage des traitements anticoagulants par les antivitamines K...).Les hémorragies intestinales associent souvent une cause locale et une cause générale et ont tendance à la répétition.Les hémorragies méningées sont les plus graves, en raison de leur localisation, et peuvent causer la mort avant toute hémostase.Les hématomes (collection de sang dans l’épaisseur des muscles, ou dans le tissu cellulaire sous-cutané) peuvent être graves en raison de leur importance (cause d’anémie aiguë) ou en raison de leur caractère compressif (compression vasculaire, nerveuse, compression des voies respiratoires). Ils conduisent le médecin à rechercher un trouble de la coagulation proprement dite et, en particulier, une hémophilie.Les hémorragies génitales , dans le sexe féminin, peuvent être sévères par leur répétition ou par leur abondance (ménométrorragies). Actuellement, on dispose d’un traitement hormonal très actif pour mettre fin à ces hémorragies (progestatifs de synthèse). Il est essentiel de faire le diagnostic entre hémorragies de cause locale ou de cause générale.Dans ce qui suit, on examinera les principaux syndromes hémorragiques constitutionnels ou acquis, classés en fonction du défaut de l’hémostase qui est à leur origine, ainsi que le principe de leur traitement.Déficit congénital de la coagulationL’hémophilie représente le type des syndromes hémorragiques constitutionnels par défaut congénital d’un facteur de coagulation. Tare récessive liée au sexe, elle frappe les sujets masculins et se transmet par les femmes qui sont cliniquement indemnes. Les premières hémorragies (ecchymoses, hématomes) apparaissent entre douze à dix-huit mois au moment de la marche; les hémorragies traumatiques sont fréquentes, et la chirurgie est dangereuse. Tous les intermédiaires existent entre les formes graves se compliquant d’hémarthroses (hémorragies intra-articulaires) touchant les grosses articulations, cause d’infirmité motrice, et les formes frustes avec temps de coagulation normal. Biologiquement, on distingue deux types d’hémophilie: l’hémophilie A (85 p. 100 des cas), due à un défaut de facteur VIII, et l’hémophilie B, due à un défaut de facteur IX. En l’absence d’un traitement curatif (qui pourrait être la thérapie génique), la thérapeutique antihémophilique ne peut qu’être substitutive, c’est-à-dire qu’elle repose sur l’apport des facteurs de coagulation qui font défaut au malade.L’utilisation, à cet effet, à partir de 1970, de fractions plasmatiques concentrées permettait d’adapter le traitement à chacun des cas d’hémophilie et d’instituer une thérapeutique préventive, en administrant ces produits de façon régulière aux patients. Le drame de la contamination virale des prélèvements sanguins dans les années 1980 a donc sévèrement touché les hémophiles. Des méthodes de chauffage, puis de purification sélective ont permis d’éliminer maintenant le risque d’infection virale des concentrés sanguins destinés aux hémophiles.L’angiohémophilie , ou maladie de Willebrand , est une affection constitutionnelle qui atteint les deux sexes. Elle comporte un allongement du temps de saignement alors que, chez l’hémophile vrai, le trouble de coagulation contraste avec un temps de saignement normal après scarification de la peau du lobule de l’oreille ou de l’avant-bras. Il existe, dans la maladie de Willebrand, un déficit en facteur VIII, mais moins important que dans l’hémophilie A. Ce déficit intéresse un profacteur VIII, car le taux de facteur VIII peut être corrigé par transfusion de sang d’un hémophile A.D’autres syndromes hémorragiques proches de l’hémophilie sont liés à un déficit congénital en un seul des facteurs de coagulation suivants: XI (PTA); V (maladie de Owren, ou hypoaccélérinémie congénitale); VII (hypoproconvertinémie congénitale); X; exceptionnellement II et XIII (cf. tableau).L’absence congénitale de fibrinogène , avec sang strictement incoagulable, est un défaut compatible avec la vie (montrant l’importance de l’hémostase plaquettaire). Des hémorragies proches de celle de l’hémophilie (hématomes extensifs) nécessitent un traitement d’urgence: transfusions de sang, de plasma ou de la fraction du plasma qui contient le fibrinogène (fraction I de Cohn).Déficits acquis de facteurs de coagulationAlors que les déficits congénitaux ne portaient que sur un seul facteur, les déficits acquis intéressent plusieurs facteurs à la fois.L’abaissement des facteurs du complexe prothrombique (dont la synthèse est faite par le foie en présence de vitamine K) dans les affections hépatobiliaires, chez le nouveau-né, et dans les intoxications par les antivitamines K (traitements anticoagulants des thromboses) est la cause d’hémorragies aisément combattues par l’administration de vitamine K1, si la cellule hépatique est indemne. Lorsque la cellule hépatique est lésée, la vitamine K1 est inefficace, et il faudra recourir à des préparations plasmatiques riches en facteurs II, VII, X et IX, désignées en France, où elles ont été préparées pour la première fois, sous le nom de PPSB (prothrombine + proconvertine + Stuart + facteur antihémophilique B). L’action de cette fraction contenant les facteurs concentrés vingt à trente fois par rapport au plasma d’origine est très efficace, aussi bien dans ces syndromes que dans l’hémophilie B.Un surdosage en héparine , au cours d’un traitement anticoagulant de thrombose, peut être la cause d’un syndrome pseudohémophilique; il est facilement traité par l’administration de sulfate de protamine.Les états de défibrination aiguë provoquent en chirurgie, et surtout en obstétrique, des accidents cataclysmiques avec sang incoagulable. Ils sont dus à une coagulation intravasculaire disséminée et se compliquent de fibrinolyse. Ils imposent un traitement d’urgence: administration de sang frais, de fibrinogène injectable. Les inhibiteurs d’enzymes autrefois préconisés tendent à faire place à un traitement prudent par l’héparine, lorsque la coagulation intravasculaire l’emporte sur la fibrinolyse.Atteintes vasculaires et plaquettairesSyndromes hémorragiques acquis d’origine vasculaireLes syndromes hémorragiques dus à une atteinte de l’hémostase primaire sont nombreux; ils sont beaucoup plus souvent acquis que constitutionnels.Parmi les atteintes purement vasculaires, figurent la tendance hémorragique constatée au cours du scorbut (avitaminose C et P) et, surtout, le purpura rhumatoïde de nature allergique ou anaphylactique, associant purpura cutané prédominant aux membres inférieurs, douleurs articulaires et signes abdominaux parfois graves (simulant une affection chirurgicale ou se compliquant réellement d’invagination, d’hémorragie digestive ou de perforation). Le pronostic est généralement bénin, sauf dans les formes abdominales et en cas d’atteinte rénale. D’autre part, ce purpura tend à récidiver au lever (purpura orthostatique). Au point de vue hématologique, les tests de l’hémostase ne sont pas perturbés; les plaquettes sont normales. La biopsie d’un élément cutané montre les capillaires dilatés et entourés de manchons inflammatoires leucocytaires.Le purpura fulminans est infiniment plus grave; il s’agit d’une capillarite suraiguë avec vastes plages ecchymotiques se nécrosant. La coagulation intravasculaire au niveau des petits vaisseaux cutanés paraît jouer un rôle important et peut justifier un traitement par l’héparine [cf. PURPURAS].Syndromes hémorragiques acquis d’origine plaquettaireLes thrombopénies (plaquettes réduites à moins de 50 000 par millimètre cube au lieu de 200 000 à 300 000), sont à l’origine de syndromes hémorragiques passagers ou durables, devenant souvent chroniques dans l’adolescence et à l’âge adulte (thrombopénie idiopathique).Les hémorragies sont essentiellement cutanées et muqueuses; les hémorragies génitales sont fréquentes et peuvent révéler la thrombopénie à la puberté. Les hémorragies méningées sont redoutables, et leur danger justifie à lui seul le recours au traitement efficace qu’est l’ablation de la rate, ou splénectomie (lorsque la thrombopénie est idiopathique et dure depuis plus de six mois à un an).Mais les thrombopénies, surtout chez l’enfant, sont souvent spontanément curables. La corticothérapie favorise la guérison en quelques semaines, que la cause première ait été infectieuse ou médicamenteuse. Les transfusions de sang ou de concentrés de plaquettes, les injections de fortes doses d’immunoglobulines par voie intraveineuse peuvent être passagèrement utilisées pour passer un cap hémorragique critique ou pour préparer à la splénectomie.Enfin, une maladie maligne du sang, telle qu’une leucémie aiguë, peut revêtir le masque d’un purpura thrombopénique, d’où la nécessité de pratiquer une ponction de moelle osseuse pour confirmer le diagnostic.On peut conclure cette brève revue des principaux syndromes hémorragiques sur l’importance de l’utilisation médicale du sang et de ses dérivés. Le sang frais, les concentrés de plaquettes, le plasma sec, les fractions du plasma (fraction I contenant le fibrinogène, cryoconcentré et concentrés de facteur VIII, fraction PPSB) permettent un traitement actif. Le choix entre ces différentes fractions concentrées nécessite un diagnostic précis fondé sur la clinique et sur les tests de l’hémostase.Autres syndromes hémorragiques constitutionnelsParmi les affections constitutionnelles, on peut mentionner la maladie de Rendu-Osler (télangiectasies hémorragiques familiales, c’est-à-dire dilatations artériolo-capillaires suivies d’une rupture des parois des vaisseaux, d’où l’apparition d’hémorragies) et les rares thrombopathies où la tendance hémorragique est due à une atteinte non pas quantitative, mais qualitative, des plaquettes (maladie de Glanzmann, maladie de Bernard-Soulier).
Dans ce qui suit, on étudiera brièvement les différents stades de la séquence aboutissant à la fibrine.ThromboplastinoformationLa transformation de la prothrombine en thrombine fait intervenir un ensemble très complexe d’activateurs (primitivement désignés sous le nom générique de thromboplastines). Elle résulte de la convergence de deux systèmes de thromboplastines différents, mais tous deux nécessaires. L’un fait intervenir des facteurs d’origine tissulaire: c’est la thromboplastinoformation extrinsèque, puisqu’elle met en jeu des éléments qui n’appartiennent pas au sang. L’autre se forme à partir d’éléments contenus dans le sang lui-même; c’est la thromboplastinoformation intrinsèque.La thromboplastinoformation intrinsèque est déclenchée par toute attrition vasculaire ou par le contact du sang avec une surface étrangère, tel le verre. Elle fait intervenir un «facteur contact» (XII), point de départ d’une véritable cascade de réactions enzymatiques dans lesquelles un facteur, une fois activé, active à son tour celui qui lui succède dans la chaîne enzymatique (fig. 1). On dit qu’un facteur s’est activé quand il acquiert une activité enzymatique spécifique. L’activation peut être de nature physique, telle celle du facteur Hageman par contact avec les surfaces «mouillables» (verre, silice...) ou de nature chimique, comme pour tous les autres facteurs de coagulation. En conclusion, à la suite de cette réaction en chaîne, le facteur X est activé.L’activation du facteur X a lieu simultanément dans l’organisme par thromboplastinoformation extrinsèque , mécanisme plus rapide que celui qui vient d’être énuméré. Celle-ci fait intervenir des sucs tissulaires à la place du facteur VIII activé. En d’autres termes, des extraits de tissus (en particulier de poumon, de cerveau, de placenta) accélèrent considérablement la coagulation. Ils sont libérés au niveau des blessures par suite de l’attrition tissulaire. Pour être actifs, ces sucs tissulaires doivent réagir avec un facteur normal du plasma, le facteur VII ou proconvertine (fig. 1). Le facteur X activé va s’associer à un autre facteur plasmatique, le facteur V (ou proaccélérine), qui se trouve activé par les premières traces de thrombine formée.ThrombinoformationLes facteurs X et V activés forment, en présence d’ions calcium et de micelles de lipides (phospholipides), un complexe doué d’une activité enzymatique, la prothrombinase, qui transforme la prothrombine en thrombine par scission protéolytique (fig. 1): c’est la thrombinoformation.La staphylocoagulase, provenant de certaines souches de staphylocoque doré, est également capable de se combiner directement avec la prothrombine humaine pour démasquer son site actif et coaguler le fibrinogène du sang.FibrinoformationLa thrombine une fois formée transforme le fibrinogène (facteur I) soluble en un réticulum de fibrine insoluble (caillot proprement dit). Les molécules de fibrinogène, qui sont très allongées, sont attaquées près de leur extrémité. Elles se trouvent ainsi amputées d’une charge négative (acides aminés diacides). Elles cessent de se repousser par des forces électrostatiques et se polymérisent, c’est-à-dire s’associent pour former un réseau solide.Le facteur XIII ou FSF, facteur de stabilisation de la fibrine, intervient pour consolider ce réseau.Inhibiteurs physiologiques de la coagulationComment expliquer que le sang coagule si rapidement dès qu’il est sorti des vaisseaux, alors que les coagulations intravasculaires spontanées sont rares? L’existence d’inhibiteurs de la coagulation est une des réponses à ce problème.Les facteurs de coagulation sont stables dans le plasma et se renouvellent avec une rapidité qui est propre à chaque facteur. Leur synthèse s’effectue au niveau du foie: elle est continue et équilibre la destruction physiologique de ces protéines très spécialisées. Lorsqu’on bloque la synthèse, la demi-vie (temps de destruction de la moitié de la quantité initialement en circulation) s’échelonne entre quatre heures, pour le facteur VII, et trois à quatre jours, pour le fibrinogène et la prothrombine.Dans les conditions physiologiques, une fois activés, les facteurs de coagulation sont très rapidement détruits au niveau du foie ou dans la circulation par des inhibiteurs, dont le principal est l’antithrombine III , capable de détruire progressivement de fortes quantités de thrombine non utilisées pour la coagulation du fibrinogène. L’héparine forme avec l’AT III un complexe qui accélère considérablement l’action anticoagulante.Fibrinolyse du caillotLa coagulation une fois terminée, le caillot se rétracte sous l’influence des plaquettes enserrées dans le réseau de fibrine. Ultérieurement, il va s’organiser; le réseau de fibrine prépare la cicatrisation en servant de matrice pour le développement de fibroblastes (tissu conjonctif).Un caillot non organisé est susceptible de se lyser c’est-à-dire de se dissoudre, à la suite de l’activation d’une enzyme fibrinolytique, appelée plasmine , qui dérive d’un précurseur inerte nommé plasminogène. Cette activation résulte de l’action d’activateurs tissulaires ou vasculaires, ou de l’urokinase, substance trouvée dans les urines normales (fig. 2).Certains streptocoques produisent un activateur du plasminogène nommé streptokinase (ou streptolysine).La plasmine est capable de scinder la fibrine en plusieurs fragments, qui agissent comme des inhibiteurs compétitifs de la transformation du fibrinogène en fibrine ou de la polymérisation de la fibrine. Il existe dans le plasma un inhibiteur de la plasmine à action progressive, l’antiplasmine, ainsi que l’alpha-2-macroglobuline qui a une action beaucoup plus rapide.2. HémorragiesLes hémorragies peuvent être de cause locale ou générale.Hémorragies de cause localeDans les hémorragies de cause locale, le mécanisme de l’hémostase n’est pas en cause; l’ensemble des défenses de l’organisme intervient en vue de remédier à la perte sanguine et de déterminer son arrêt. Une plaie artérielle avec issue en saccades de sang rouge vif impose souvent une intervention d’urgence: pose d’un garrot, compression de l’artère en amont, suture ou ligature chirurgicale, faute de quoi un état d’anémie aiguë se constitue très rapidement, caractérisant le choc hémorragique, avec pâleur, perte de conscience, pouls petit et rapide, effondrement de la tension artérielle.De telles hémorragies se voient non seulement dans les sections accidentelles d’artères, au cours de traumatismes de la route ou du travail, mais également dans certaines affections obstétricales (grossesse extra-utérine) où une artère peut être érodée. C’est aussi le cas des grandes hémoptysies (crachements de sang faisant irruption dans les voies aériennes), et des grandes hématémèses (vomissements de sang), qui compliquent certains ulcères d’estomac creusant en profondeur, ou certaines cirrhoses avec hypertension portale (rupture de varices œsophagiennes nécessitant la pose d’un ballon œsophagogastrique gonflable).Dans tous ces cas d’hémorragies dramatiques de cause locale, deux gestes s’imposent: l’hémostase locale (souvent chirurgicale), et la transfusion massive de sang.Hémorragies de cause généralePrincipales localisationsLes hémorragies cutanées , pétéchies (petites hémorragies cutanées de la taille de confettis, qui surviennent spontanément sous forme de «purpura»), et ecchymoses (classiques «bleus») se rencontrent aussi bien dans les purpuras d’origine vasculaire ou plaquettaire, que dans les atteintes de la coagulation proprement dite.Les hémorragies muqueuses comprennent les épistaxis (saignement de nez), les gingivorragies (saignement des gencives), les hémorragies après extractions dentaires (l’extraction dentaire révèle des tendances hémorragiques demeurées souvent latentes; en effet, il y a véritable arrachement d’un paquet vasculonerveux sans hémostase locale).Les hématuries , ou hémorragies urinaires, se rencontrent aussi bien dans les atteintes rénales que dans les syndromes hémorragiques des purpuras ou des troubles de la coagulation (hémophilie, surdosage des traitements anticoagulants par les antivitamines K...).Les hémorragies intestinales associent souvent une cause locale et une cause générale et ont tendance à la répétition.Les hémorragies méningées sont les plus graves, en raison de leur localisation, et peuvent causer la mort avant toute hémostase.Les hématomes (collection de sang dans l’épaisseur des muscles, ou dans le tissu cellulaire sous-cutané) peuvent être graves en raison de leur importance (cause d’anémie aiguë) ou en raison de leur caractère compressif (compression vasculaire, nerveuse, compression des voies respiratoires). Ils conduisent le médecin à rechercher un trouble de la coagulation proprement dite et, en particulier, une hémophilie.Les hémorragies génitales , dans le sexe féminin, peuvent être sévères par leur répétition ou par leur abondance (ménométrorragies). Actuellement, on dispose d’un traitement hormonal très actif pour mettre fin à ces hémorragies (progestatifs de synthèse). Il est essentiel de faire le diagnostic entre hémorragies de cause locale ou de cause générale.Dans ce qui suit, on examinera les principaux syndromes hémorragiques constitutionnels ou acquis, classés en fonction du défaut de l’hémostase qui est à leur origine, ainsi que le principe de leur traitement.Déficit congénital de la coagulationL’hémophilie représente le type des syndromes hémorragiques constitutionnels par défaut congénital d’un facteur de coagulation. Tare récessive liée au sexe, elle frappe les sujets masculins et se transmet par les femmes qui sont cliniquement indemnes. Les premières hémorragies (ecchymoses, hématomes) apparaissent entre douze à dix-huit mois au moment de la marche; les hémorragies traumatiques sont fréquentes, et la chirurgie est dangereuse. Tous les intermédiaires existent entre les formes graves se compliquant d’hémarthroses (hémorragies intra-articulaires) touchant les grosses articulations, cause d’infirmité motrice, et les formes frustes avec temps de coagulation normal. Biologiquement, on distingue deux types d’hémophilie: l’hémophilie A (85 p. 100 des cas), due à un défaut de facteur VIII, et l’hémophilie B, due à un défaut de facteur IX. En l’absence d’un traitement curatif (qui pourrait être la thérapie génique), la thérapeutique antihémophilique ne peut qu’être substitutive, c’est-à-dire qu’elle repose sur l’apport des facteurs de coagulation qui font défaut au malade.L’utilisation, à cet effet, à partir de 1970, de fractions plasmatiques concentrées permettait d’adapter le traitement à chacun des cas d’hémophilie et d’instituer une thérapeutique préventive, en administrant ces produits de façon régulière aux patients. Le drame de la contamination virale des prélèvements sanguins dans les années 1980 a donc sévèrement touché les hémophiles. Des méthodes de chauffage, puis de purification sélective ont permis d’éliminer maintenant le risque d’infection virale des concentrés sanguins destinés aux hémophiles.L’angiohémophilie , ou maladie de Willebrand , est une affection constitutionnelle qui atteint les deux sexes. Elle comporte un allongement du temps de saignement alors que, chez l’hémophile vrai, le trouble de coagulation contraste avec un temps de saignement normal après scarification de la peau du lobule de l’oreille ou de l’avant-bras. Il existe, dans la maladie de Willebrand, un déficit en facteur VIII, mais moins important que dans l’hémophilie A. Ce déficit intéresse un profacteur VIII, car le taux de facteur VIII peut être corrigé par transfusion de sang d’un hémophile A.D’autres syndromes hémorragiques proches de l’hémophilie sont liés à un déficit congénital en un seul des facteurs de coagulation suivants: XI (PTA); V (maladie de Owren, ou hypoaccélérinémie congénitale); VII (hypoproconvertinémie congénitale); X; exceptionnellement II et XIII (cf. tableau).L’absence congénitale de fibrinogène , avec sang strictement incoagulable, est un défaut compatible avec la vie (montrant l’importance de l’hémostase plaquettaire). Des hémorragies proches de celle de l’hémophilie (hématomes extensifs) nécessitent un traitement d’urgence: transfusions de sang, de plasma ou de la fraction du plasma qui contient le fibrinogène (fraction I de Cohn).Déficits acquis de facteurs de coagulationAlors que les déficits congénitaux ne portaient que sur un seul facteur, les déficits acquis intéressent plusieurs facteurs à la fois.L’abaissement des facteurs du complexe prothrombique (dont la synthèse est faite par le foie en présence de vitamine K) dans les affections hépatobiliaires, chez le nouveau-né, et dans les intoxications par les antivitamines K (traitements anticoagulants des thromboses) est la cause d’hémorragies aisément combattues par l’administration de vitamine K1, si la cellule hépatique est indemne. Lorsque la cellule hépatique est lésée, la vitamine K1 est inefficace, et il faudra recourir à des préparations plasmatiques riches en facteurs II, VII, X et IX, désignées en France, où elles ont été préparées pour la première fois, sous le nom de PPSB (prothrombine + proconvertine + Stuart + facteur antihémophilique B). L’action de cette fraction contenant les facteurs concentrés vingt à trente fois par rapport au plasma d’origine est très efficace, aussi bien dans ces syndromes que dans l’hémophilie B.Un surdosage en héparine , au cours d’un traitement anticoagulant de thrombose, peut être la cause d’un syndrome pseudohémophilique; il est facilement traité par l’administration de sulfate de protamine.Les états de défibrination aiguë provoquent en chirurgie, et surtout en obstétrique, des accidents cataclysmiques avec sang incoagulable. Ils sont dus à une coagulation intravasculaire disséminée et se compliquent de fibrinolyse. Ils imposent un traitement d’urgence: administration de sang frais, de fibrinogène injectable. Les inhibiteurs d’enzymes autrefois préconisés tendent à faire place à un traitement prudent par l’héparine, lorsque la coagulation intravasculaire l’emporte sur la fibrinolyse.Atteintes vasculaires et plaquettairesSyndromes hémorragiques acquis d’origine vasculaireLes syndromes hémorragiques dus à une atteinte de l’hémostase primaire sont nombreux; ils sont beaucoup plus souvent acquis que constitutionnels.Parmi les atteintes purement vasculaires, figurent la tendance hémorragique constatée au cours du scorbut (avitaminose C et P) et, surtout, le purpura rhumatoïde de nature allergique ou anaphylactique, associant purpura cutané prédominant aux membres inférieurs, douleurs articulaires et signes abdominaux parfois graves (simulant une affection chirurgicale ou se compliquant réellement d’invagination, d’hémorragie digestive ou de perforation). Le pronostic est généralement bénin, sauf dans les formes abdominales et en cas d’atteinte rénale. D’autre part, ce purpura tend à récidiver au lever (purpura orthostatique). Au point de vue hématologique, les tests de l’hémostase ne sont pas perturbés; les plaquettes sont normales. La biopsie d’un élément cutané montre les capillaires dilatés et entourés de manchons inflammatoires leucocytaires.Le purpura fulminans est infiniment plus grave; il s’agit d’une capillarite suraiguë avec vastes plages ecchymotiques se nécrosant. La coagulation intravasculaire au niveau des petits vaisseaux cutanés paraît jouer un rôle important et peut justifier un traitement par l’héparine [cf. PURPURAS].Syndromes hémorragiques acquis d’origine plaquettaireLes thrombopénies (plaquettes réduites à moins de 50 000 par millimètre cube au lieu de 200 000 à 300 000), sont à l’origine de syndromes hémorragiques passagers ou durables, devenant souvent chroniques dans l’adolescence et à l’âge adulte (thrombopénie idiopathique).Les hémorragies sont essentiellement cutanées et muqueuses; les hémorragies génitales sont fréquentes et peuvent révéler la thrombopénie à la puberté. Les hémorragies méningées sont redoutables, et leur danger justifie à lui seul le recours au traitement efficace qu’est l’ablation de la rate, ou splénectomie (lorsque la thrombopénie est idiopathique et dure depuis plus de six mois à un an).Mais les thrombopénies, surtout chez l’enfant, sont souvent spontanément curables. La corticothérapie favorise la guérison en quelques semaines, que la cause première ait été infectieuse ou médicamenteuse. Les transfusions de sang ou de concentrés de plaquettes, les injections de fortes doses d’immunoglobulines par voie intraveineuse peuvent être passagèrement utilisées pour passer un cap hémorragique critique ou pour préparer à la splénectomie.Enfin, une maladie maligne du sang, telle qu’une leucémie aiguë, peut revêtir le masque d’un purpura thrombopénique, d’où la nécessité de pratiquer une ponction de moelle osseuse pour confirmer le diagnostic.On peut conclure cette brève revue des principaux syndromes hémorragiques sur l’importance de l’utilisation médicale du sang et de ses dérivés. Le sang frais, les concentrés de plaquettes, le plasma sec, les fractions du plasma (fraction I contenant le fibrinogène, cryoconcentré et concentrés de facteur VIII, fraction PPSB) permettent un traitement actif. Le choix entre ces différentes fractions concentrées nécessite un diagnostic précis fondé sur la clinique et sur les tests de l’hémostase.Autres syndromes hémorragiques constitutionnelsParmi les affections constitutionnelles, on peut mentionner la maladie de Rendu-Osler (télangiectasies hémorragiques familiales, c’est-à-dire dilatations artériolo-capillaires suivies d’une rupture des parois des vaisseaux, d’où l’apparition d’hémorragies) et les rares thrombopathies où la tendance hémorragique est due à une atteinte non pas quantitative, mais qualitative, des plaquettes (maladie de Glanzmann, maladie de Bernard-Soulier).
Encyclopédie Universelle. 2012.